Artificial intelligence (AI) has solved one of biology’s grand challenges
“What the DeepMind team has managed to achieve is fantastic and will change the future of structural biology and protein research,” says Janet Thornton, director emeritus of the European Bioinformatics Institute. “This is a 50-year-old problem,” adds John Moult, a structural biologist at the University of Maryland, Shady Grove, and co-founder of the competition, Critical Assessment of Protein Structure Prediction (CASP). “I never thought I’d see this in my lifetime.”
The human body uses tens of thousands of different proteins, each a string of dozens to many hundreds of amino acids. The order of those amino acids dictates how the myriad pushes and pulls between them give rise to proteins’ complex 3D shapes, which, in turn, determine how they function. Knowing those shapes helps researchers devise drugs that can lodge in proteins’ pockets and crevices. And being able to synthesize proteins with a desired structure could speed the development of enzymes that make biofuels and degrade waste plastic.
For decades, researchers deciphered proteins’ 3D structures using experimental techniques such as x-ray crystallography or cryo–electron microscopy (cryo-EM). But such methods can take months or years and don’t always work. Structures have been solved for only about 170,000 of the more than 200 million proteins discovered across life forms.
In the 1960s, researchers realized if they could work out all individual interactions within a protein’s sequence, they could predict its 3D shape. With hundreds of amino acids per protein and numerous ways each pair of amino acids can interact, however, the number of possible structures per sequence was astronomical. Computational scientists jumped on the problem, but progress was slow.
In 1994, Moult and colleagues launched CASP, which takes place every 2 years. Entrants get amino acid sequences for about 100 proteins whose structures are not known. Some groups compute a structure for each sequence, while other groups determine it experimentally. The organizers then compare the computational predictions with the lab results and give the predictions a global distance test (GDT) score. Scores above 90 on the zero to 100 scale are considered on par with experimental methods, Moult says.
Even in 1994, predicted structures for small, simple proteins could match experimental results. But for larger, challenging proteins, computations’ GDT scores were about 20, “a complete catastrophe,” says Andrei Lupas, a CASP judge and evolutionary biologist at the Max Planck Institute for Developmental Biology. By 2016, competing groups had reached scores of about 40 for the hardest proteins, mostly by drawing insights from known structures of proteins that were closely related to the CASP targets.
When DeepMind first competed in 2018, its algorithm, called AlphaFold, relied on this comparative strategy. But AlphaFold also incorporated a computational approach called deep learning, in which the software is trained on vast data troves—in this case, the sequences and structures of known proteins—and learns to spot patterns. DeepMind won handily, beating the competition by an average of 15% on each structure, and winning GDT scores of up to about 60 for the hardest targets.
But the predictions were still too coarse to be useful, says John Jumper, who heads AlphaFold’s development at DeepMind. “We knew how far we were from biological relevance.” To do better, Jumper and his colleagues combined deep learning with an “attention algorithm” that mimics the way a person might assemble a jigsaw puzzle: first connecting pieces in small clumps—in this case clusters of amino acids—and then searching for ways to join the clumps in a larger whole. Working with a computer network built around 128 machine learning processors, they trained the algorithm on all 170,000 or so known protein structures.
And it worked. Across target proteins in this year’s CASP, AlphaFold achieved a median GDT score of 92.4. For the most challenging proteins, AlphaFold scored a median of 87, 25 points above the next best predictions. It even excelled at solving structures of proteins that sit wedged in cell membranes, which are central to many human diseases but notoriously difficult to solve with x-ray crystallography. Venki Ramakrishnan, a structural biologist at the Medical Research Council Laboratory of Molecular Biology, calls the result “a stunning advance on the protein folding problem.”
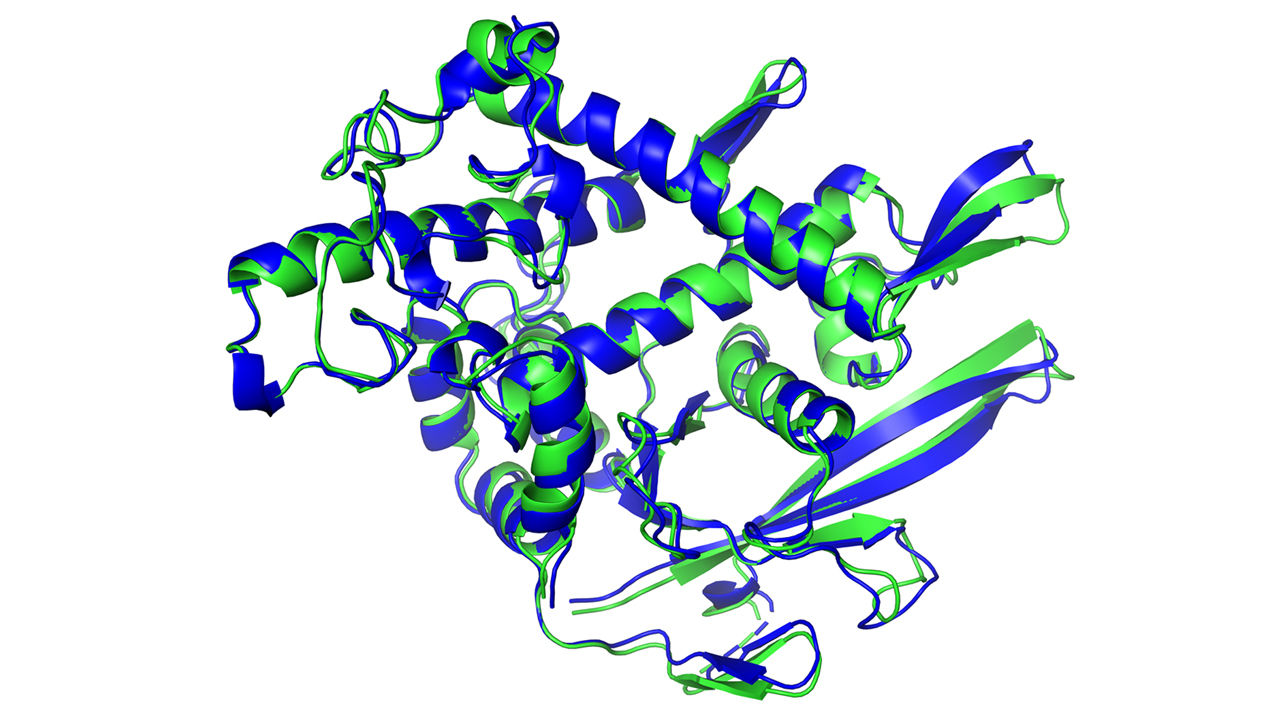
All of the groups in this year’s competition improved, Moult says. But with AlphaFold, Lupas says, “The game has changed.” The organizers even worried DeepMind may have been cheating somehow. So Lupas set a special challenge: a membrane protein from a species of archaea, an ancient group of microbes. For 10 years, his research team tried every trick in the book to get an x-ray crystal structure of the protein. “We couldn’t solve it.”
But AlphaFold had no trouble. It returned a detailed image of a three-part protein with two long helical arms in the middle. The model enabled Lupas and his colleagues to make sense of their x-ray data; within half an hour, they had fit their experimental results to AlphaFold’s predicted structure. “It’s almost perfect,” Lupas says. “They could not possibly have cheated on this. I don’t know how they do it.”
As a condition of entering CASP, DeepMind—like all groups—agreed to reveal sufficient details about its method for other groups to re-create it. That will be a boon for experimentalists, who will be able to use accurate structure predictions to make sense of opaque x-ray and cryo-EM data. It could also enable drug designers to quickly work out the structure of every protein in new and dangerous pathogens like SARS-CoV-2, a key step in the hunt for molecules to block them, Moult says.
Still, AlphaFold doesn’t do everything well yet. In the contest, it faltered noticeably on one protein, an amalgam of 52 small repeating segments, which distort each others’ positions as they assemble. Jumper says the team now wants to train AlphaFold to solve such structures, as well as those of complexes of proteins that work together to carry out key functions in the cell.
Even though one grand challenge has fallen, others will undoubtedly emerge. “This isn’t the end of something,” Thornton says. “It’s the beginning of many new things.”
Robert F. Service